Molecular Docking with GNINA 1.0
David Ryan Koes
Royal Society of Chemistry Chemical Information & Computer Applications Group
David Ryan Koes
Royal Society of Chemistry Chemical Information & Computer Applications Group
May 27, 2021


Get Started: https://colab.research.google.com/drive/1GXmk1v8C-c4UtyKFqIm9HnsrVYH0pI-c
%%html
<style>
div.prompt {display:none}
div.output_subarea {max-width: 100%}
</style>
<script>
$3Dmolpromise = new Promise((resolve, reject) => {
require(['https://3dmol.org/build/3Dmol-nojquery.js'], function(){
resolve();});
});
require(['https://cdnjs.cloudflare.com/ajax/libs/Chart.js/2.2.2/Chart.js'], function(Ch){
Chart = Ch;
});
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
//the callback is provided a canvas object and data
var chartmaker = function(canvas, labels, data) {
var ctx = $(canvas).get(0).getContext("2d");
var dataset = {labels: labels,
datasets:[{
data: data,
backgroundColor: "rgba(150,64,150,0.5)",
fillColor: "rgba(150,64,150,0.8)",
}]};
var myBarChart = new Chart(ctx,{type:'bar',data:dataset,options:{legend: {display:false},
scales: {
yAxes: [{
ticks: {
min: 0,
}
}]}}});
};
$(".input .o:contains(html)").closest('.input').hide();
</script>
<script src="https://bits.csb.pitt.edu/asker.js/lib/asker.js"></script>
Acknowledgements¶


Andrew McNutt, Paul Francoeur, Rishal Aggarwal, Tomohide Masuda, Rocco Meli, Matthew Ragoza, Jocelyn Sunseri
%%html
<div id="whydock" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#whydock';
jQuery(divid).asker({
id: divid,
question: "Why do you most want to dock?",
answers: ['Predict pose','Virtual screening','Affinity prediction',"I don't know"],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".input .o:contains(html)").closest('.input').hide();
</script>
What is molecular docking?¶
Predict the most likely conformation and pose of a ligand in a protein binding site.
- Sample conformational space
- Score poses
- Ideally score equals affinity or can be used to productively rank compounds
- Score $\ne$ Free Energy
%%html
<iframe width="560" height="315" src="https://3dmol.org/tests/docking.html" title="docking" frameborder="0" allow="accelerometer; autoplay; clipboard-write; encrypted-media; gyroscope; picture-in-picture" allowfullscreen></iframe>
<script>
$(".input .o:contains(html)").closest('.input').hide();
</script>
Inherent limitations of docking¶
Docking is intended to be high-throughput and fundamentally limiting approximations are made to achieve this.
- Receptor usually kept rigid or mostly rigid (limited side-chain flexibility)
- Ligand flexibility usually limited to torsions
- No explicit solvent model
Software Lineage¶

AutoDock Vina¶
Designed and implemented by Dr. Oleg Trott at the Scripps Research Institute.
Shared no code with AutoDock.
Focus on performance. Created new scoring function optimized for pose prediction.
Open Source Apache License
Published 2009, last update (version 1.1.2) 2011
Software Lineage¶

smina
Scoring and minimization with AutoDock Vina
We forked Vina to make it easier to use, especially for custom scoring function development and ligand minimization.
(Almost) identical behavior as Autodock Vina (just easier to use).
Apache/GPL2 Open Source License
Very stable source code. In maintence mode. Features are a subset of GNINA.

!wget https://downloads.sourceforge.net/project/smina/smina.static
!wget https://github.com/gnina/gnina/releases/download/v1.0.1/gnina
!du -sh smina.static gnina
I wasn't kidding about the extra dependencies!
However, if you are going to use gnina frequently you should build it from source so it uses the versions of libraries installed on your system (especially CUDA) which will result in a much smaller executable.
!du -sh /usr/local/bin/gnina
Running GNINA¶
!chmod +x ./gnina #make executable
!./gnina
%%html
<div id="gnsucc" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#gnsucc';
jQuery(divid).asker({
id: divid,
question: "Were you able to run gnina in colab?",
answers: ['Yes','No','Eh'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".input .o:contains(html)").closest('.input').hide();
</script>
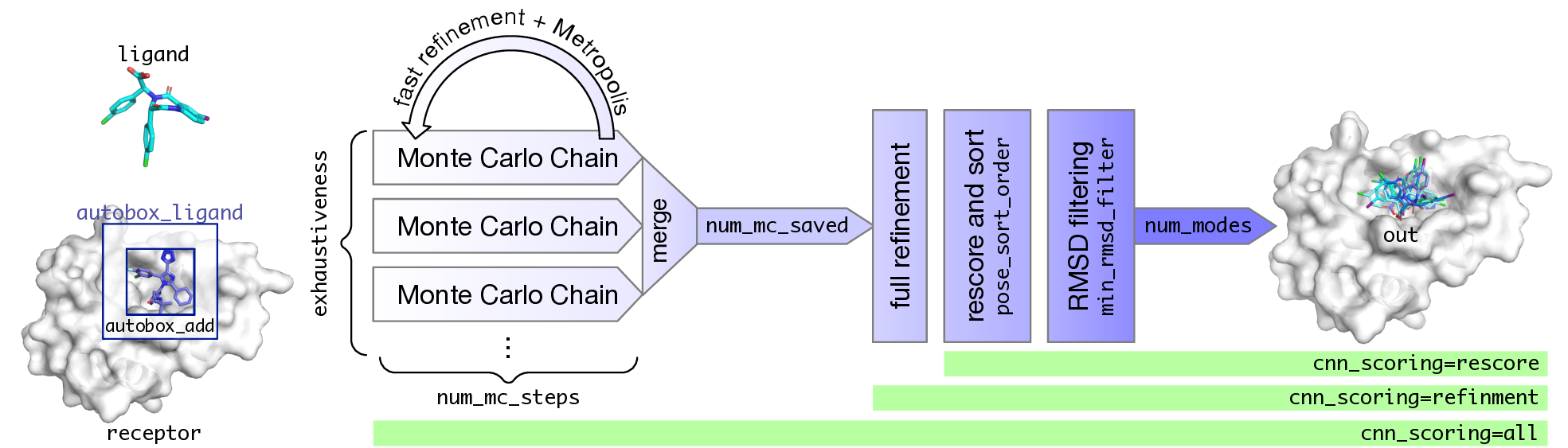
How does it work?¶
Setup Example¶
!wget http://files.rcsb.org/download/3ERK.pdb
!grep ATOM 3ERK.pdb > rec.pdb
!obabel rec.pdb -Orec.pdb # "sanitizing" receptor for openbabel
!grep SB4 3ERK.pdb > lig.pdb
import py3Dmol
v = py3Dmol.view(height=400)
v.addModel(open('rec.pdb').read())
v.setStyle({'cartoon':{},'stick':{'radius':0.15}})
v.addModel(open('lig.pdb').read())
v.setStyle({'model':1},{'stick':{'colorscheme':'greenCarbon'}})
v.zoomTo({'model':1})
Protein Preparation¶
Any file format supported by Open Babel is acceptable. Every atom in the provided file will be treated as part of the receptor.
Check for
- missing atoms
- alternative residues
- co-factors
Receptors already in a bound conformation are best, but remember to remove the ligand.
Protonation
- By default Open Babel will be used to infer protonation
- Generally only adds hydrogens
- To see what protonation will be used:
obabel rec.pdb -h -xr -Orec.pdbqt
- If PDBQT file is provided it will be taken as is with no hydrogens changed.
Ligand Preparation¶
Any file format supported by Open Babel is acceptable.
Need valid 3D conformation
Only torsions (rotatable bonds) are sampled during docking
- Ring conformations and stereoisomers are not sampled
!obabel -:'C1CNCCC1n1cnc(c2ccc(cc2)F)c1c1ccnc(n1)N' -Ol2.sdf --gen2D
This is not a valid ligand conformation (but you will still be able to dock it).
v = py3Dmol.view(height=300)
v.addModel(open('l2.sdf').read())
v.setStyle({'stick':{'colorscheme':'greenCarbon'}})
v.zoomTo()
!obabel -:'C1CNCCC1n1cnc(c2ccc(cc2)F)c1c1ccnc(n1)N' -Ol3.sdf --gen3D
v = py3Dmol.view(height=400)
v.addModel(open('l3.sdf').read())
v.setStyle({'stick':{'colorscheme':'greenCarbon'}})
v.zoomTo()
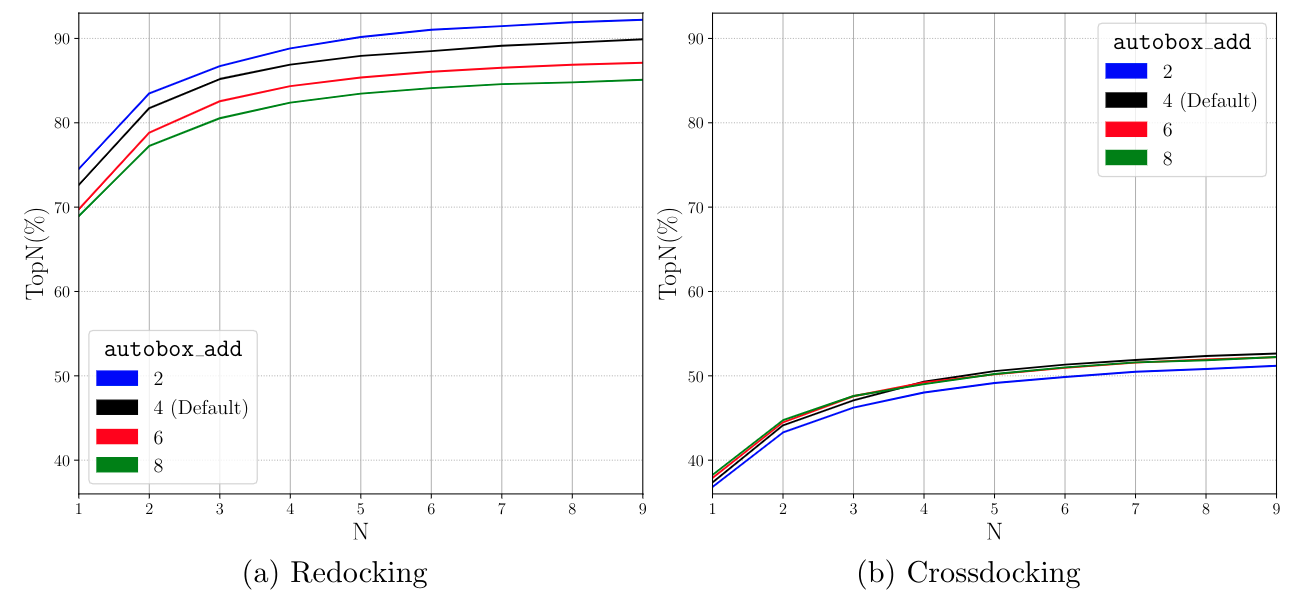
Defining the Binding Site¶
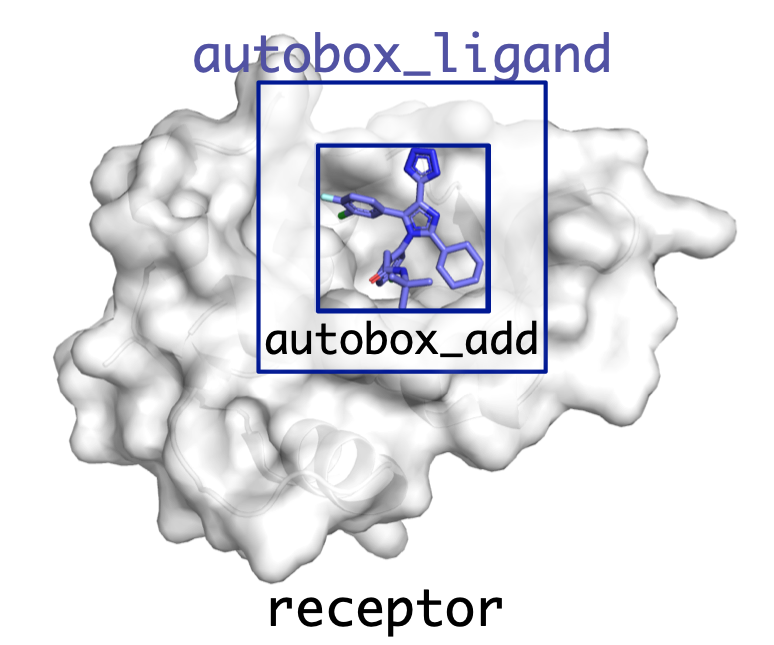
All poses are sampled within a box defined by the user.
Can be specified manually (--center_x, --size_x, etc.) but typically much easier to provide an autobox ligand.
A box is created that exactly inscribes the atom coordinates of the provided ligand and then is expanded by autobox_add (default 4Å) in every dimension. If needed so provide enough room for the ligand to freely rotate the box is then further extended (autobox_extend).
Can provide any molecule for autobox_ligand (e.g. binding pocket residues, fpocket alpha spheres).
%%html
<div id="autobox" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#autobox';
jQuery(divid).asker({
id: divid,
question: "What do you think happens to docking performance when autobox_add is increased?",
answers: ['Docking is slower but better','Docking is faster and better','Docking is slower and worse','Docking is faster but worse'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".input .o:contains(html)").closest('.input').hide();
</script>
Let's Dock!¶
!./gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb
Two improvements:
- Set the random seed for reproducibility (on same system)
- Specify an output file so generated poses are saved
!./gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb --seed 0 -o docked.sdf.gz
How good are the results?
We'll measure RMSD of poses with obrms from Open Babel which you can install in colab with:
!apt install openbabel
!obrms --firstonly lig.pdb docked.sdf.gz
import gzip
v = py3Dmol.view(height=400)
v.addModel(open('rec.pdb').read())
v.setStyle({'cartoon':{},'stick':{'radius':.1}})
v.addModel(open('lig.pdb').read())
v.setStyle({'model':1},{'stick':{'colorscheme':'dimgrayCarbon','radius':.125}})
v.addModelsAsFrames(gzip.open('docked.sdf.gz','rt').read())
v.setStyle({'model':2},{'stick':{'colorscheme':'greenCarbon'}})
v.animate({'interval':1000}); v.zoomTo({'model':1}); v.rotate(90)
%%html
<div id="betterl" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#betterl';
jQuery(divid).asker({
id: divid,
question: "If we dock a generated conformer (l3.sdf) instead, what happens to the RMSD?",
answers: ['Better','Same-ish','Worse'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".input .o:contains(html)").closest('.input').hide();
</script>
!./gnina -r rec.pdb -l l3.sdf --autobox_ligand lig.pdb --seed 0 -o docked.sdf.gz
!obrms --firstonly lig.pdb docked.sdf.gz
Sampling¶
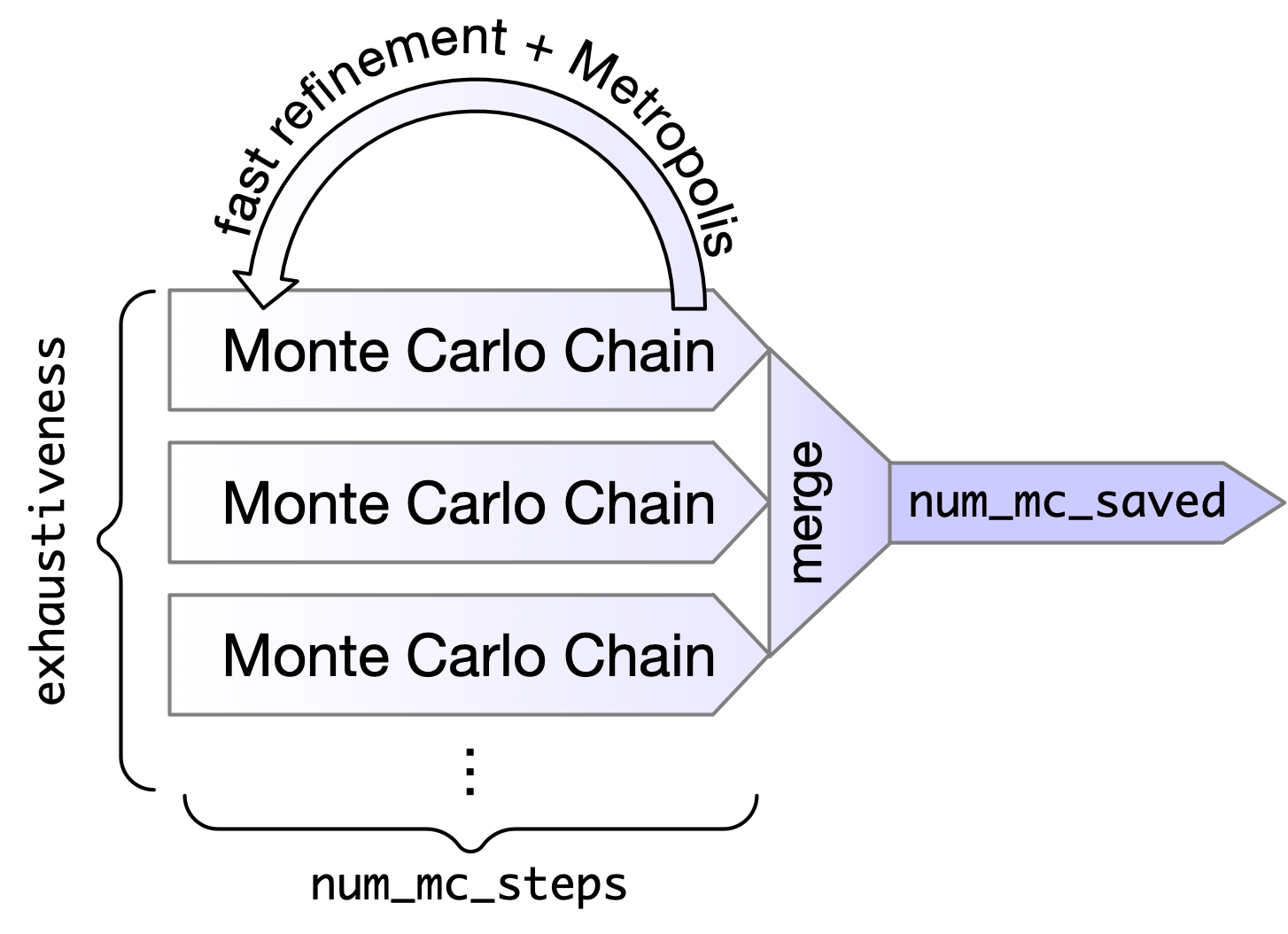
- Degrees of freedom
- 6 rigid body motions (x,y,z,pitch,yaw,roll)
- Internal torsions (not other angles/bond lengths)
- Initially randomize all degrees of freedom
- no bias to starting conformation DoF
- is biased by non-DoF conformations (e.g. ring pucker)
- Monte Carlo Chain
- Apply a random transformation (translation, rotation, or torsion)
- Perform fast refinement (truncated BFGS) of result with "soft" potentials
- Metropolis criterion to accept result as new conformation
- The more change improves conformation, more likely it is selected
- Best scoring conformations are retained.
Sampling¶
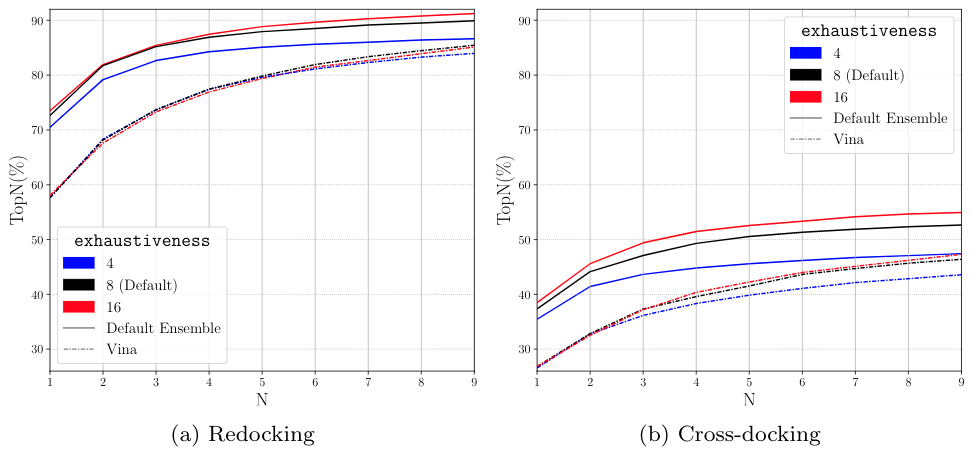
--exhaustivenessThe number of MC chains. These can be done in parallel. This is the recommended way to change the amount of sampling.--num_mc_stepsHow many iterations each MC chain performs. By default is heuristically scaled based on number of degrees of freedom (more flexible ligands will take longer). Don't recommend using unless you want to do a "quick and dirty" docking run.--num_mc_savedNumber of best scoring conformations retained by each chain and overall process. Default is max of 50 or the number of requested output conformations. Shouldn't have to change this.
Timing exhaustiveness¶
%%time
!./gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb --seed 0 --exhaustiveness 1 > /dev/null 2>&1
MC chains are run in parallel so increasing exhaustivess won't be much slower as long as there are enough cores
%%time
!./gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb --seed 0 --exhaustiveness 4 > /dev/null 2>&1
But if MC chains can't run in parallel expect a roughly linear increase in time.
%%time
!./gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb --seed 0 --exhaustiveness 4 --cpu 1 > /dev/null 2>&1
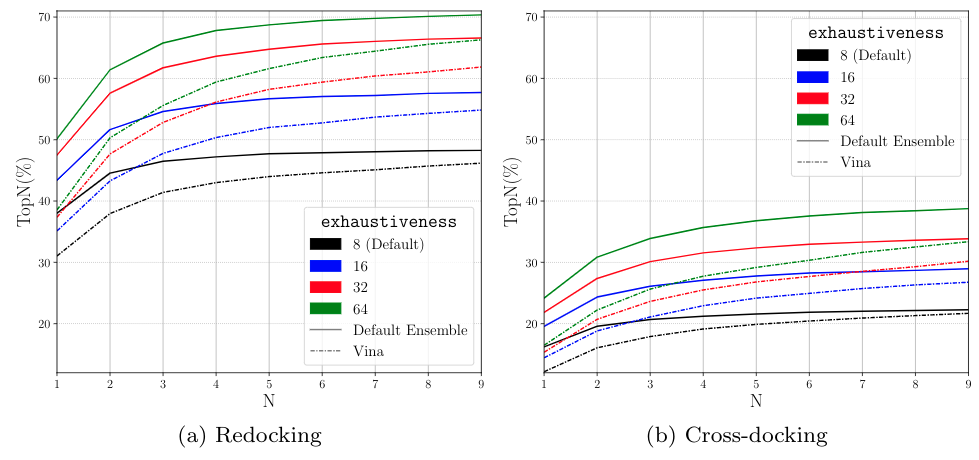
For a typical docking run, there are diminishing returns in increasing the exhaustivness and the default (8) is sufficient.
Scoring¶
Empirical (e.g. Vina)
- Fast and interprettable
- A collection of weighted terms
- By default used for search and refinement
CNN
- Slower (especially without GPU) but more predictive
- By default used only for final ranking
--cnn_scoring=rescore
AutoDock Vina Scoring¶

There is no electrostatic term. Partial charges are not used. Electrostatic interactions are accounted for with hydrogen bond term.
Metals are modeled as hydrogen donors.
Terms were selected and parameterized for pose prediction performance (both speed and quality).
Final scoring function was then linearly reweighted to fit score to free energies (kcal/mol).
!./gnina --score_only -r rec.pdb -l lig.pdb --verbosity=2
Alternative Empirical Scoring¶
!./gnina --help | grep scoring | head -3
- default/vina AutoDock Vina
- vinardo A reparameterized Vina (https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0155183)
- ad4_scoring A reimplmentation of AutoDock4 scoring (includes electrostatics and solvation)
- ignore the rest
Custom Empirical Scoring¶
Scoring functions can be defined in text files by parameterizing built-in terms
!./gnina --print_terms
Example¶
Create a file of equally-weight terms. Firt column is weight. Second the parameterized term. Remainder ignored.
open('everything.txt','wt').write('''
1.0 ad4_solvation(d-sigma=3.6,_s/q=0.01097,_c=8) desolvation, s/q is charge dependence
1.0 ad4_solvation(d-sigma=3.6,_s/q=0.0,_c=8)
1.0 electrostatic(i=1,_^=100,_c=8) i is the exponent of the distance, see everything.h for details
1.0 electrostatic(i=2,_^=100,_c=8)
1.0 gauss(o=0,_w=0.5,_c=8) o is offset, w is width of gaussian
1.0 gauss(o=3,_w=2,_c=8)
1.0 repulsion(o=0,_c=8) o is offset of squared distance repulsion
1.0 hydrophobic(g=0.5,_b=1.5,_c=8) g is a good distance, b the bad distance
1.0 non_hydrophobic(g=0.5,_b=1.5,_c=8) value is linearly interpolated between g and b
1.0 vdw(i=4,_j=8,_s=0,_^=100,_c=8) i and j are LJ exponents
1.0 vdw(i=6,_j=12,_s=1,_^=100,_c=8) s is the smoothing, ^ is the cap
1.0 non_dir_h_bond(g=-0.7,_b=0,_c=8) good and bad
1.0 non_dir_anti_h_bond_quadratic(o=0.4,_c=8) like repulsion, but for hbond, don't use
1.0 non_dir_h_bond_lj(o=-0.7,_^=100,_c=8) LJ 10-12 potential, capped at ^
1.0 acceptor_acceptor_quadratic(o=0,_c=8) quadratic potential between hydrogen bond acceptors
1.0 donor_donor_quadratic(o=0,_c=8) quadratic potential between hydroben bond donors
1.0 num_tors_div div constant terms are not linearly independent
1.0 num_heavy_atoms_div
1.0 num_heavy_atoms these terms are just added
1.0 num_tors_add
1.0 num_tors_sqr
1.0 num_tors_sqrt
1.0 num_hydrophobic_atoms
1.0 ligand_length
''');
Example¶
--custom_scoring will replace empirical scoring with function defined in provided file.
!./gnina -r rec.pdb -l lig.pdb --score_only --custom_scoring everything.txt
Hacky Use Case¶
We wanted to soft "covalently" dock a ligand. Modified system to change atom types of bonding atoms to Chlorine and Sulfur (non-physical modification) and used this custom scoring function:
-0.035579 gauss(o=0,_w=0.5,_c=8)
-0.005156 gauss(o=3,_w=2,_c=8)
0.840245 repulsion(o=0,_c=8)
-0.035069 hydrophobic(g=0.5,_b=1.5,_c=8)
-0.587439 non_dir_h_bond(g=-0.7,_b=0,_c=8)
1.923 num_tors_div
-100.0 atom_type_gaussian(t1=Chlorine,t2=Sulfur,o=0,_w=3,_c=8)CNN Scoring¶
Convolutional neural networks learn spatially related features of an input grid to generate a prediction.
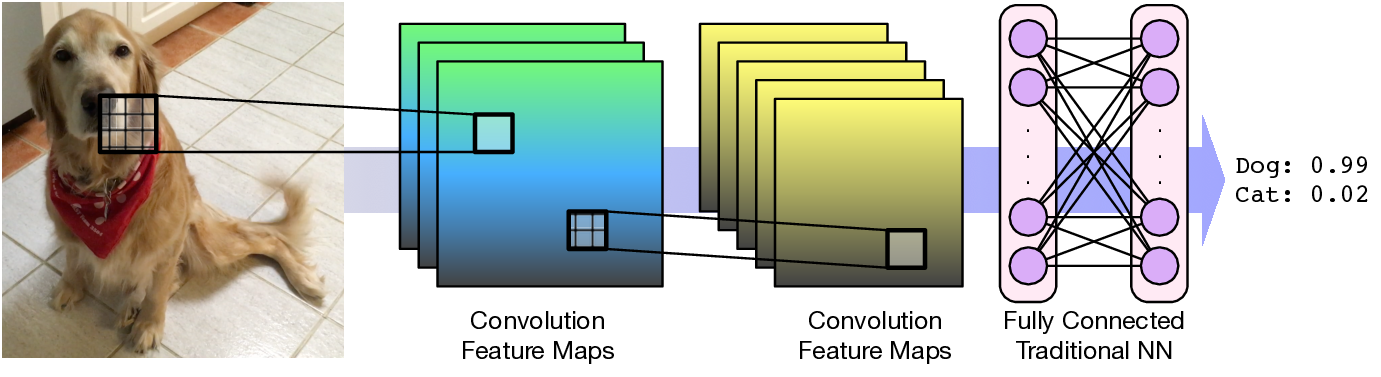
CNN Scoring¶
Atoms are represented as Gaussian densities on a 24Å grid. There is a separate channel for each atom type.
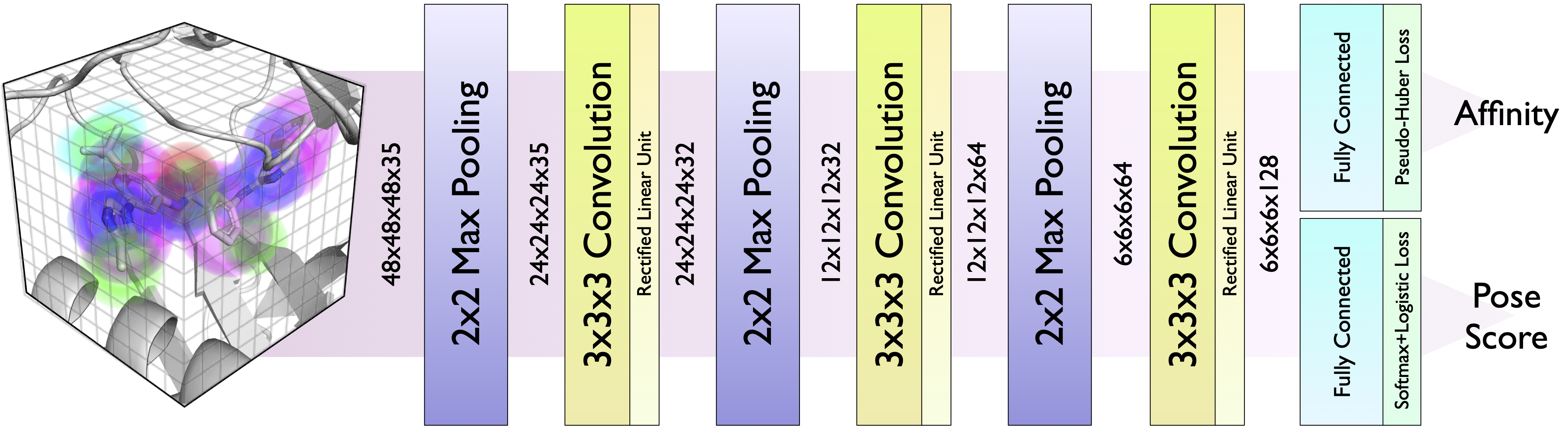
CNN Models¶
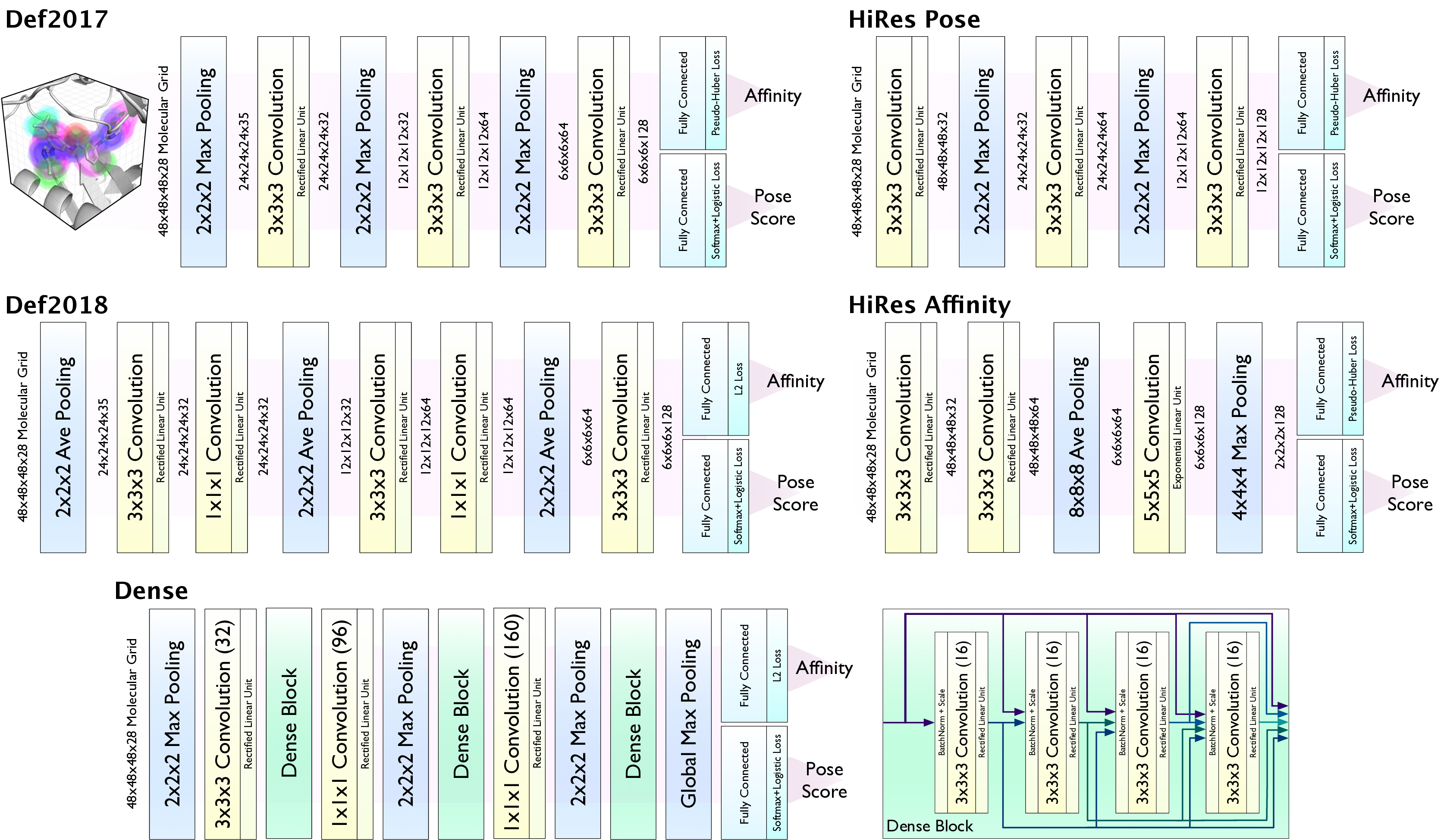
CNN Model Ensembles¶
The default is to use an ensemble of 5 models that was found to have the best performance.
!./gnina --help | grep "cnn arg" -A 12
A CNN model predicts both pose quality (CNNScore) and binding affinity (CNNaffinity).
!./gnina --score_only -r rec.pdb -l lig.pdb | grep CNN
CNNscore is a probability that the pose is a "good" (<2 RMSD) pose
CNNaffnity is predicted affinity in "pK" units - 1$\mu M$ is 6, 1$nM$ is 9
CNNvariance is the variance of predicted affinities across the ensemble. It is not a score, but a measure of uncertainty (lower is better).
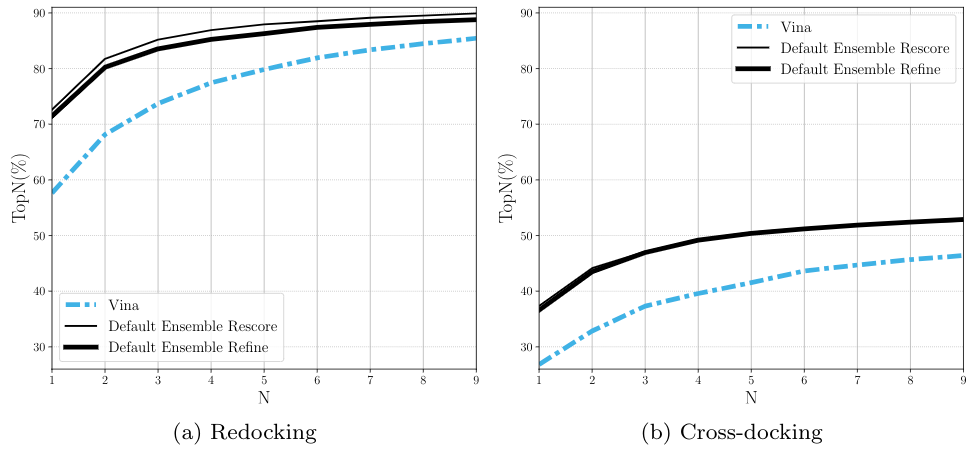
CNN Scoring Performance¶
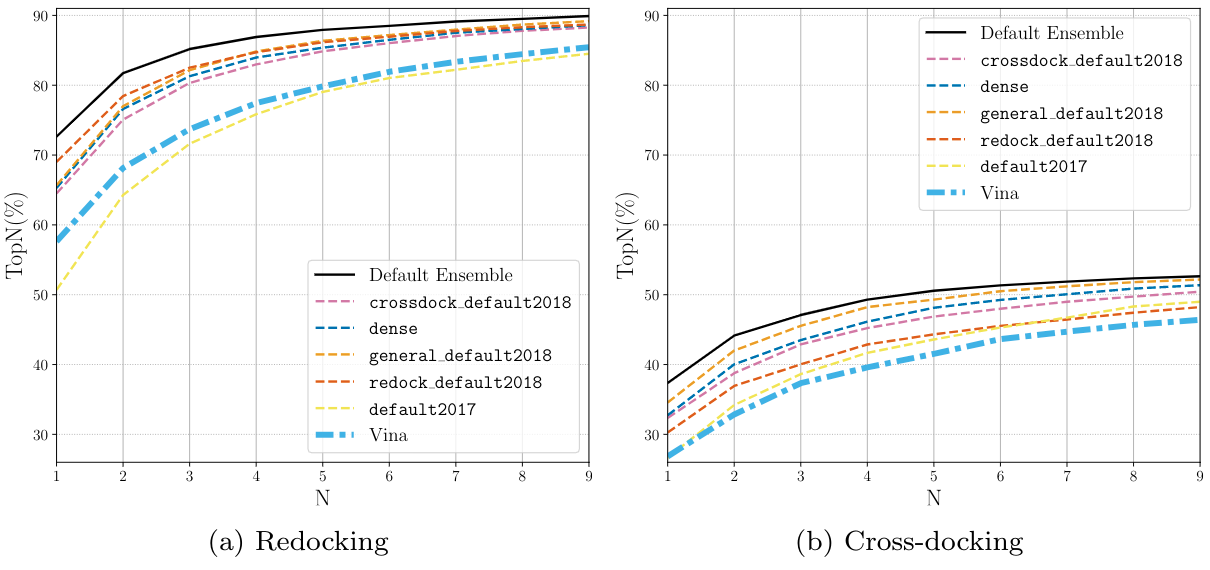
Ranking¶
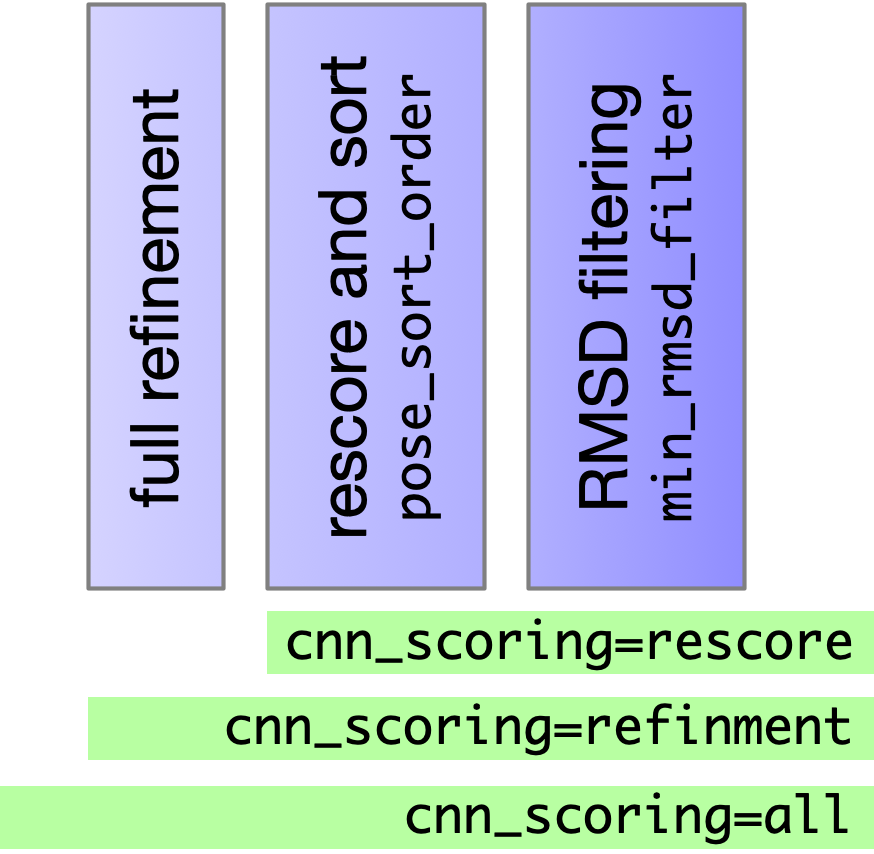
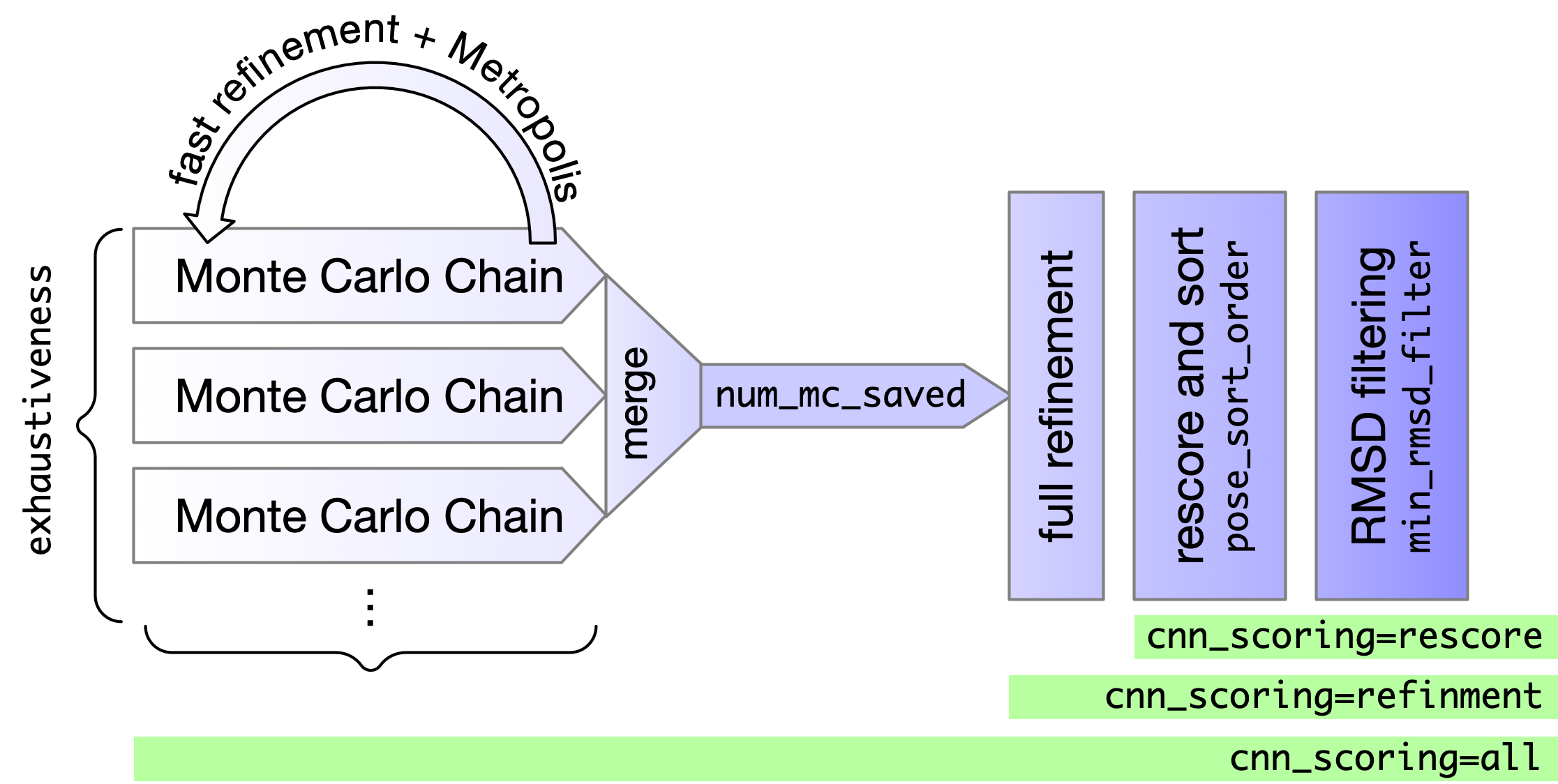
- Top
num_mc_savedposes from sampling are refined (BFGS) with full (not soft) potentials - Resulting poses are rescored and sorted according to
--pose_sort_order--pose_sort_order=CNNscore(default) Poses with highest probability of being low RMSD according to CNN are ranked highest--pose_sort_order=CNNaffinityPoses with highest CNN predicted binding affinity are ranked highest--pose_sort_order=EnergyPoses with lowest Vina predicted energy are ranked highest
- Final ranked list is filtered to remove poses within
--min_rmsd_filter(default 1Å)
Note: Changing the sort order can change what poses are returned, not just their ordering.
Using CNN for refinment (--cnn_scoring=refinement) is not helpful and is much slower.
CNN scoring is slow without a GPU. Any modern NVIDIA GPU with $\ge$4GB RAM should work.
%%time
!./gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb --seed 0 > /dev/null 2>&1
%%time
!CUDA_VISIBLE_DEVICES= ./gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb --seed 0
Whole Protein Docking¶
Set the receptor to the autobox_ligand.
!./gnina -r rec.pdb -l lig.pdb --autobox_ligand rec.pdb -o wdocking.sdf.gz --seed 0
v = py3Dmol.view(height=400)
v.addModel(open('rec.pdb').read())
v.setStyle({'cartoon':{},'stick':{'radius':.1}})
v.addModel(open('lig.pdb').read())
v.setStyle({'model':1},{'stick':{'colorscheme':'dimgrayCarbon','radius':.125}})
v.addModelsAsFrames(gzip.open('wdocking.sdf.gz','rt').read())
v.setStyle({'model':2},{'stick':{'colorscheme':'greenCarbon'}})
v.animate({'interval':1000}); v.zoomTo(); v.rotate(90)
We do not see diminishing returns when increasing exhaustiveness with whole protein docking.
!wget http://files.rcsb.org/download/4ERK.pdb
!grep ATOM 4ERK.pdb > rec2.pdb
!obabel rec2.pdb -Orec2.pdb
!grep OLO 4ERK.pdb > lig2.pdb
Let's dock the ligand from 3ERK to the 4ERK structure.
!./gnina -r rec2.pdb -l lig.pdb --autobox_ligand lig2.pdb --seed 0 -o 3erk_to_4erk.sdf.gz
!obrms --firstonly lig.pdb 3erk_to_4erk.sdf.gz
v = py3Dmol.view(height=380)
v.addModel(open('3ERK.pdb').read())
v.setStyle({'model':0},{'cartoon':{'colorscheme':'greenCarbon'},'stick':{'radius':.1,'colorscheme':'greenCarbon'}})
v.addModel(open('4ERK.pdb').read())
v.setStyle({'model':1},{'cartoon':{'colorscheme':'yellowCarbon'},'stick':{'radius':.1,'colorscheme':'yellowCarbon'}})
v.addModel(gzip.open('3erk_to_4erk.sdf.gz','rt').read())
v.setStyle({'model':2},{'stick':{'colorscheme':'magentaCarbon'}})
v.zoomTo({'model':2})
Flexible Docking¶
--flexProvide flexible side-chains as PDBQT file. Rigid part of receptor should have these side-chains removed.--flexresSpecify side-chains by comma separated list of chain:resid Recommended--flexdistAll side-chains with atoms this distance fromflexdist_ligandwill be set as flexible.--flexdist_ligandLigand to use to identify side-chains by distance.--flex_limitHard limit on number of flexible residues--flex_maxSoft limit on number of flexible residues (only closest are kept)--out_flexFile to write flexible side-chain output to.makeflex.pyis provided to reassemble into full structures.
Let's try to improve our docking by making side-chains within 3Å of cognate ligand flexible.
!./gnina -r rec2.pdb -l lig.pdb --autobox_ligand lig2.pdb --seed 0 -o flexdocked.sdf.gz --flexdist 4 --flexdist_ligand lig2.pdb --out_flex flexout.pdb
!obrms --firstonly lig.pdb flexdocked.sdf.gz
v = py3Dmol.view(height=340,width=940)
v.addModel(open('3ERK.pdb').read())
v.setStyle({'model':0},{'cartoon':{'colorscheme':'greenCarbon'},'stick':{'radius':.1,'colorscheme':'greenCarbon'}})
v.addModel(open('rec2.pdb').read())
v.setStyle({'model':1},{'cartoon':{'colorscheme':'yellowCarbon'},'stick':{'radius':.1,'colorscheme':'yellowCarbon'}})
v.addModel(gzip.open('flexdocked.sdf.gz','rt').read())
v.setStyle({'model':2},{'stick':{'colorscheme':'magentaCarbon'}})
v.addModel(open('flexout.pdb').read())
v.setStyle({'model':3},{'stick':{'colorscheme':'magentaCarbon'}})
v.zoomTo({'model':2}); v.rotate(90,'x')
Can do slightly better by being selective of what residues to make flexible and increasing exhaustiveness.
!./gnina -r rec2.pdb -l lig.pdb --autobox_ligand lig2.pdb --seed 0 -o flexdocked2.sdf.gz --exhaustiveness 16 --flexres A:52,A:103 --out_flex flexout2.pdb
!obrms --firstonly lig.pdb flexdocked2.sdf.gz
v = py3Dmol.view(height=340,width=940)
v.addModel(open('3ERK.pdb').read())
v.setStyle({'model':0},{'cartoon':{'colorscheme':'greenCarbon'},'stick':{'radius':.1,'colorscheme':'greenCarbon'}})
v.addModel(open('rec2.pdb').read())
v.setStyle({'model':1},{'cartoon':{'colorscheme':'yellowCarbon'},'stick':{'radius':.1,'colorscheme':'yellowCarbon'}})
v.addModel(gzip.open('flexdocked2.sdf.gz','rt').read())
v.setStyle({'model':2},{'stick':{'colorscheme':'magentaCarbon'}})
v.addModel(open('flexout2.pdb').read())
v.setStyle({'model':3},{'stick':{'colorscheme':'magentaCarbon'}})
v.zoomTo({'model':2}); v.rotate(90,'x')
Flexible Docking Recommendations¶
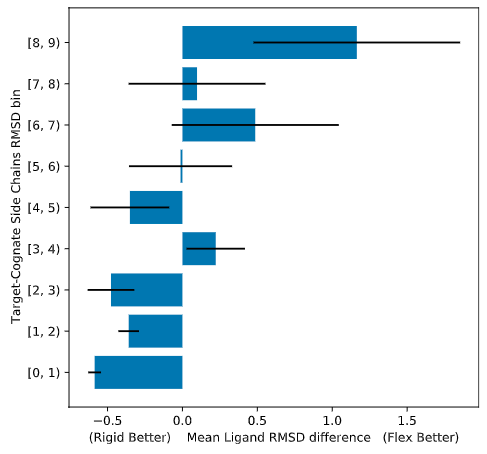
- Usually not worth it
- Increasing degrees of freedom increases false positives
- If you have an ensemble of bound protein conformations, use that
- includes backbone flexibility
- Can be useful for targetting a small number of known flexible side-chains
Virtual Screening¶
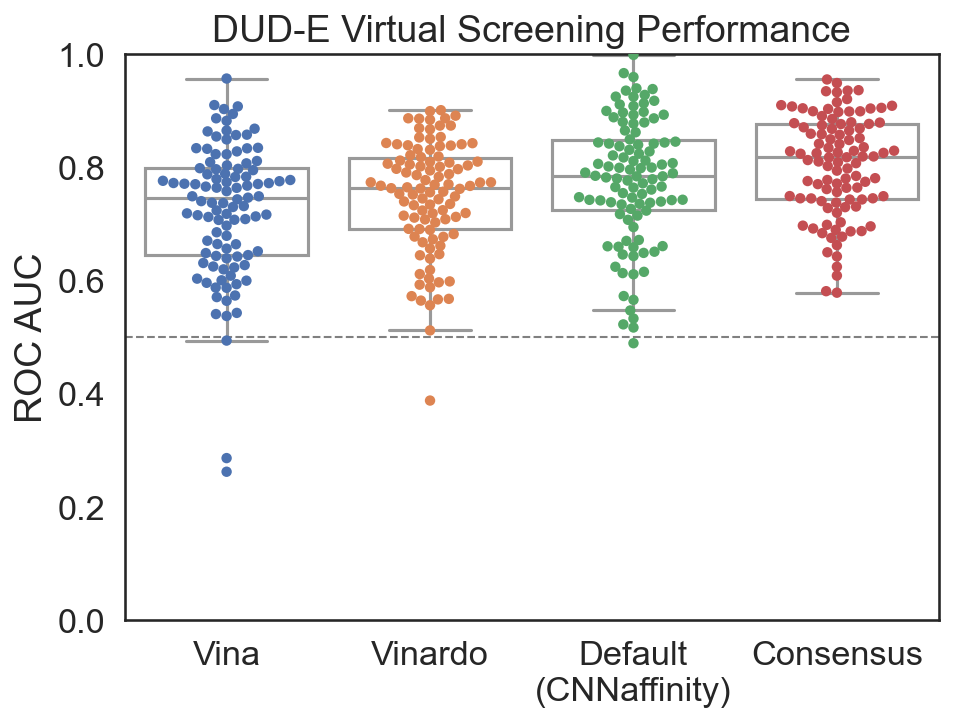
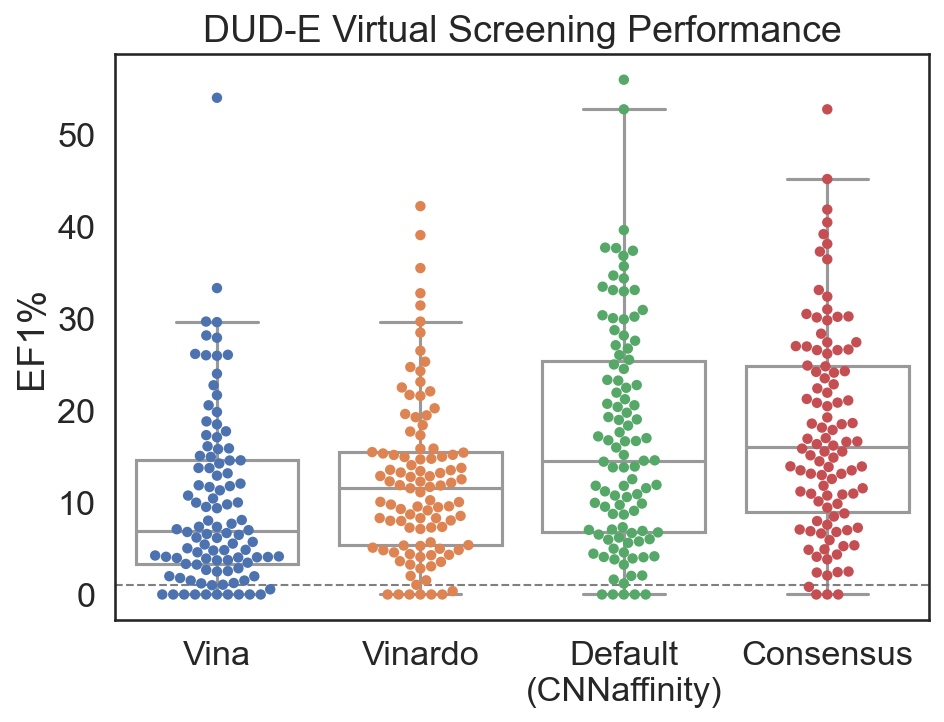
High-throughput screening recommendations¶
- Pre-filter library by molecular properties
- Remove highly flexible ligands
- Carefully manage cpu usage (
--cpu$\le$--exhaustiveness) - It is okay to share a GPU, but may be memory limited
- Avoid unnecessary receptor processing
- Provide as PDBQT
- Dock multiple ligands per a run (multi-ligand input)
- Don't do it
The Alternative: Pharmacophore Search¶
Advantages¶
- Uses expert human insight to define query
- Fast (millions of molecules in seconds)
- Results are already "docked"
- Can still using GNINA scoring to optimize/rank hits
Disadvantages¶
- Relies on expert human insight to define query
- Especially difficult when no bound ligand
- Exploration of alternative binding modes requires multiple queries
%%html
<div id="pharmexp" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#pharmexp';
jQuery(divid).asker({
id: divid,
question: "Have you done a pharmacophore search?",
answers: ['Yes w/Pharmit','Yes','No'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".input .o:contains(html)").closest('.input').hide();
</script>
Pharmit: Interactive Exploration of Chemical Space¶
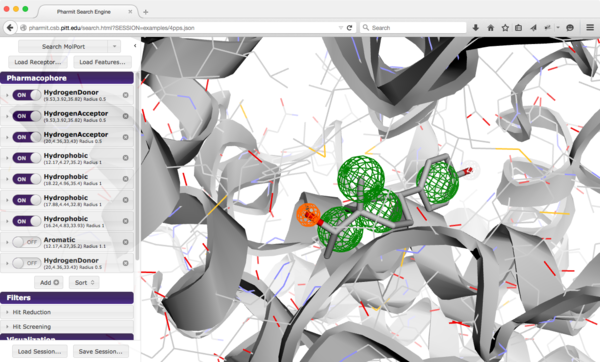
Key Points¶
- Pharmit stores rigid conformers in a special index that allows rapid pharmacophore search of millions of structures
- Several libraries with millions of commercially available compounds
- Can also create your own libraries
- Pharmacophore query specifies the 3D arrangement of essential interaction features
- Pharmacophore query specifies what must be present, not what shouldn't be
- Shape constraints can filter severe steric clashes
- Recommend minimum filtering by molecular properties
- Minimization refines ligand pose and provides Vina-based ranking
- mRMSD is how much the ligand has changed from pharmacophore-aligned pose
- large values imply no longer matches pharmacophore
- mRMSD is how much the ligand has changed from pharmacophore-aligned pose
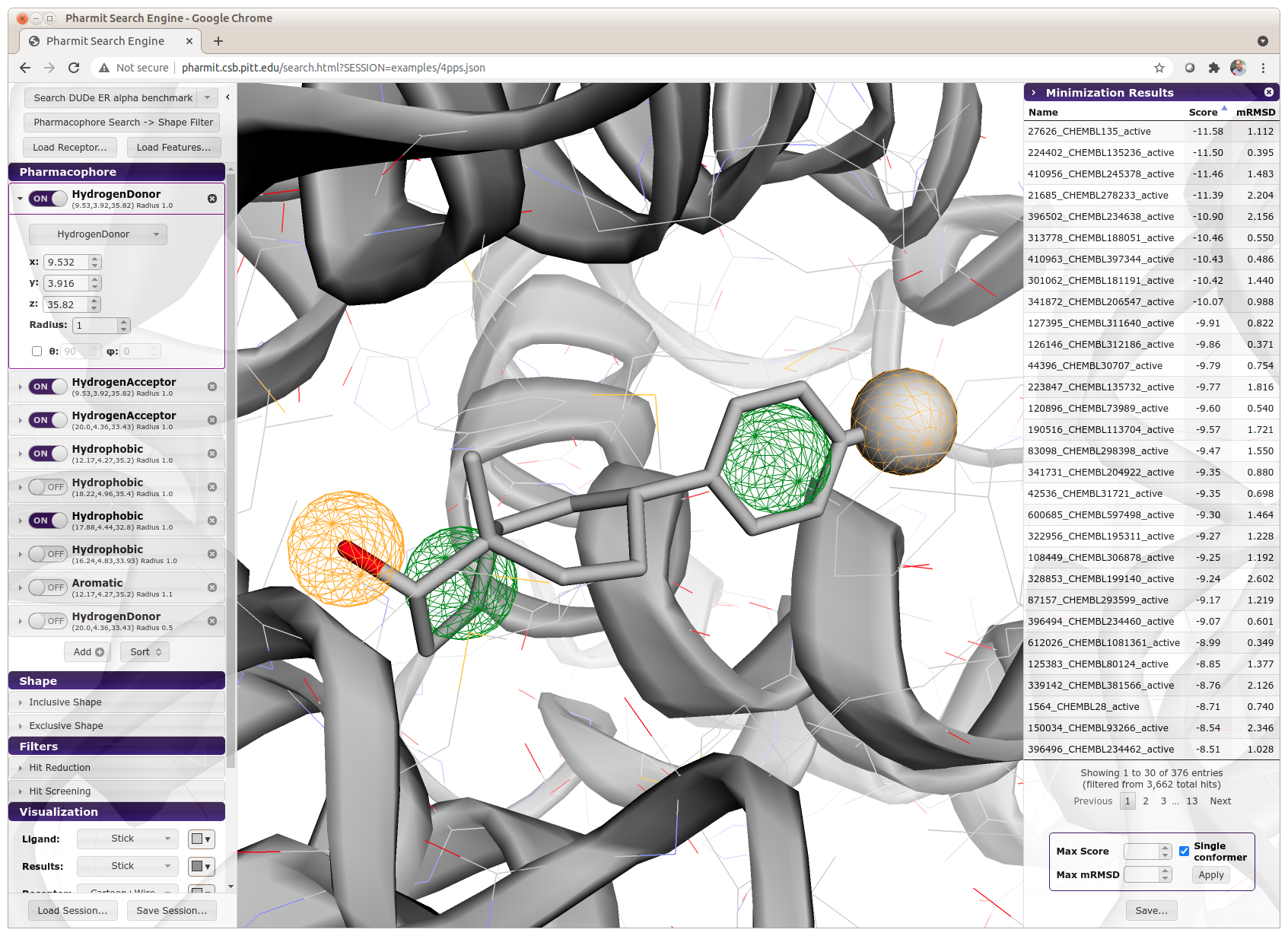
Rescoring Pharmit Results¶
Get receptor...
!wget http://files.rcsb.org/download/4PPS.pdb
!grep ^ATOM 4PPS.pdb > errec.pdb
Rescoring Pharmit Results¶
!./gnina -r errec.pdb -l minimized_results.sdf.gz --minimize -o gnina_scored.sdf.gz --scoring vinardo
Process Scores¶
When the output is an sdf file, scores are included as data fields.
from openbabel import pybel
import pandas as pd
scores = []
for mol in pybel.readfile('sdf','gnina_scored.sdf.gz'):
scores.append({'title': mol.title,
'CNNscore': float(mol.data['CNNscore']),
'CNNaffinity': float(mol.data['CNNaffinity']),
'Vinardo': float(mol.data['minimizedAffinity'])})
scores = pd.DataFrame(scores)
scores['label'] = scores.title.str.contains('active')
scores
from sklearn.metrics import roc_auc_score, roc_curve, auc
import matplotlib.pyplot as plt
%matplotlib inline
plt.figure(dpi=150)
plt.plot([0,1],[0,1],'k--',alpha=0.5,linewidth=1)
fpr,tpr,_ = roc_curve(scores.label,-scores.Vinardo)
plt.plot(fpr,tpr,label="Vinardo (AUC = %.2f)"%auc(fpr,tpr))
fpr,tpr,_ = roc_curve(scores.label,scores.CNNaffinity)
plt.plot(fpr,tpr,label="CNNaffinity (AUC = %.2f)"%auc(fpr,tpr))
fpr,tpr,_ = roc_curve(scores.label, scores.CNNaffinity.rank() + (-scores.Vinardo).rank())
plt.plot(fpr,tpr,label="Consensus (AUC = %.2f)"%auc(fpr,tpr))
plt.legend(loc='lower right')
plt.xlabel('False Positive Rate')
plt.ylabel('True Positive Rate')
plt.gca().set_aspect('equal')

Custom Scoring with GNINA Descriptors¶
!gnina -r errec.pdb -l minimized_results.sdf.gz --score_only --custom_scoring everything.txt > scores.txt 2>&1
!head -30 scores.txt
import subprocess, io, re
terms = pd.read_csv(io.BytesIO(subprocess.check_output("grep \#\# scores.txt | sed 's/## //'",shell=True)),delim_whitespace=True)
terms[['CNNscore','CNNaffinity','CNNvariance']] = re.findall(r'CNNscore: (\S+)\s*CNNaffinity: (\S+)\s*CNNvariance: (\S+)',open('scores.txt').read())
terms['label'] = terms.Name.str.contains('active')
terms
import sklearn
from sklearn.linear_model import *
X = terms.drop(['Name','label'],axis=1).astype(float) # features
Y = terms.label
X
model = LogisticRegression(solver='liblinear')
cvpredict = sklearn.model_selection.cross_val_predict(model, X, Y, method='predict_proba')
fpr,tpr,_ = roc_curve(Y,cvpredict[:,1])
fig,ax = plt.subplots(1,1,dpi=100)
ax.plot(fpr,tpr,label="Combined CV ROC (AUC=%.2f)"%auc(fpr,tpr))
plt.xlabel('False Positive Rate')
plt.ylabel('True Positive Rate')
plt.legend()
ax.set_aspect('equal');

%%html
<div id="mpredict" style="width: 500px"></div>
<script>
$('head').append('<link rel="stylesheet" href="https://bits.csb.pitt.edu/asker.js/themes/asker.default.css" />');
var divid = '#mpredict';
jQuery(divid).asker({
id: divid,
question: "Would this model be useful for further screening?",
answers: ['Yes','No','Eh'],
server: "https://bits.csb.pitt.edu/asker.js/example/asker.cgi",
charter: chartmaker})
$(".input .o:contains(html)").closest('.input').hide();
</script>
Skepticism is always warranted with learned models¶

- Should rigorously cross-validate (e.g. scaffold split)
- Need training sets that accurately represent screening library
- Actives and decoys should not be trivially seperable
- Structure-based models should be pose-sensitive
- Interrogate model (easy with empirical features)
Do not use virtual screening classification models to dock/minimize!¶
Training CNN Models¶
Beyond the scope of this workshop (sorry!)
Scripts and documentation here: https://github.com/gnina/scripts
Need to get data into a text file with this format:
<label> <affinity> <receptor> <ligand>Then design and train models in Caffe.
